
 French
French Deutsch
DeutschOrganophosphorus chemistry
Organophosphorus chemistry is the scientific study of the synthesis and properties of organophosphorus compounds, which are organic compounds containing phosphorus.[1] They are used primarily in pest control as an alternative to chlorinated hydrocarbons that persist in the environment. Some organophosphorus compounds are highly effective insecticides, although some are extremely toxic to humans, including sarin and VX nerve agents.[2]
Phosphorus, like nitrogen, is in group 15 of the periodic table, and thus phosphorus compounds and nitrogen compounds have many similar properties.[3][4][5] The definition of organophosphorus compounds is variable, which can lead to confusion. In industrial and environmental chemistry, an organophosphorus compound need contain only an organic substituent, but need not have a direct phosphorus-carbon (P-C) bond.[citation needed] Thus a large proportion of pesticides (e.g., malathion), are often included in this class of compounds.
Phosphorus can adopt a variety of oxidation states, and it is general to classify organophosphorus compounds based on their being derivatives of phosphorus(V) vs phosphorus(III), which are the predominant classes of compounds. In a descriptive but only intermittently used nomenclature, phosphorus compounds are identified by their coordination number σ and their valency λ. In this system, a phosphine is a σ3λ3 compound.
Organophosphorus(V) compounds, main categories
[edit]Phosphate esters and amides
[edit]Phosphate esters have the general structure P(=O)(OR)3 feature P(V). Such species are of technological importance as flame retardant agents, and plasticizers. Lacking a P−C bond, these compounds are in the technical sense not organophosphorus compounds but esters of phosphoric acid. Many derivatives are found in nature, such as phosphatidylcholine. Phosphate ester are synthesized by alcoholysis of phosphorus oxychloride. A variety of mixed amido-alkoxo derivatives are known, one medically significant example being the anti-cancer drug cyclophosphamide. Also derivatives containing the thiophosphoryl group (P=S) include the pesticide malathion. The organophosphates prepared on the largest scale are the zinc dithiophosphates, as additives for motor oil. Several million kilograms of this coordination complex are produced annually by the reaction of phosphorus pentasulfide with alcohols.[6]

Phosphoryl thioates are thermodynamically much stabler than thiophosphates, which can rearrange at high temperature or with a catalytic alkylant to the former:[7]: 73–76
- SP(OR)3 → OP(OR)2SR
In the environment, all these phosphorus(V) compounds break down via hydrolysis to eventually afford phosphate and the organic alcohol or amine from which they are derived.
Phosphonic and phosphinic acids and their esters
[edit]Phosphonates are esters of phosphonic acid and have the general formula RP(=O)(OR')2. Phosphonates have many technical applications, a well-known member being glyphosate, better known as Roundup. With the formula (HO)2P(O)CH2NHCH2CO2H, this derivative of glycine is one of the most widely used herbicides. Bisphosphonates are a class of drugs to treat osteoporosis. The nerve gas agent sarin, containing both C–P and F–P bonds, is a phosphonate.[citation needed]
Phosphinates feature two P–C bonds, with the general formula R2P(=O)(OR'). A commercially significant member is the herbicide glufosinate. Similar to glyphosate mentioned above, it has the structure CH3P(O)(OH)CH2CH2CH(NH2)CO2H.
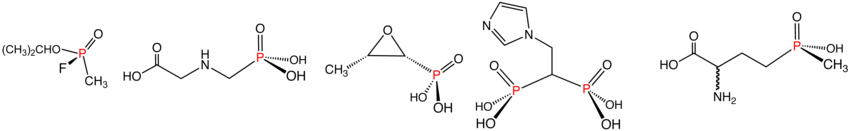
Illustrative examples of phosphonates and phosphinates in the order shown: Sarin (phosphonate), Glyphosate (phosphonate), fosfomycin (phosphonate), zoledronic acid (phosphonate), and Glufosinate (phosphinate). In aqueous solution, phosphonic acids ionize to give the corresponding organophosphonates.

The Michaelis–Arbuzov reaction is the main method for the synthesis of these compounds. For example, dimethylmethylphosphonate (see figure above) arises from the rearrangement of trimethylphosphite, which is catalyzed by methyl iodide. In the Horner–Wadsworth–Emmons reaction and the Seyferth–Gilbert homologation, phosphonates are used in reactions with carbonyl compounds. The Kabachnik–Fields reaction is a method for the preparation of aminophosphonates. These compounds contain a very inert bond between phosphorus and carbon. Consequently, they hydrolyze to give phosphonic and phosphinic acid derivatives, but not phosphate.[citation needed]
Phosphine oxides, imides, and chalcogenides
[edit]Phosphine oxides (designation σ4λ5) have the general structure R3P=O with formal oxidation state V. Phosphine oxides form hydrogen bonds and some are therefore soluble in water. The P=O bond is very polar with a dipole moment of 4.51 D for triphenylphosphine oxide.[citation needed]
Compounds related to phosphine oxides include phosphine imides (R3PNR') and related chalcogenides (R3PE, where E = S, Se, Te). These compounds are some of the most thermally stable organophosphorus compounds. In general, they are less basic than the corresponding phosphine oxides, which can adduce to thiophosphoryl halides:[7]: 73
- R3PO + X3PS → R3P+–O–P+X2–S− + X−
Some phosphorus sulfides can undergo a reverse Arbuzov rearrangement to a dialkylthiophosphinate ester.[7]: 55
Phosphonium salts and phosphoranes
[edit]Compounds with the formula [PR4+]X− comprise the phosphonium salts. These species are tetrahedral phosphorus(V) compounds. From the commercial perspective, the most important member is tetrakis(hydroxymethyl)phosphonium chloride, [P(CH2OH)4]Cl, which is used as a fire retardant in textiles. Approximately 2M kg are produced annually of the chloride and the related sulfate.[6] They are generated by the reaction of phosphine with formaldehyde in the presence of the mineral acid:
- PH3 + HX + 4 CH2O → [P(CH2OH)4+]X−
A variety of phosphonium salts can be prepared by alkylation and arylation of organophosphines:
- PR3 + R'X → [PR3R'+]X−
The methylation of triphenylphosphine is the first step in the preparation of the Wittig reagent.

Illustrative phosphorus(V) compounds: the phosphonium ion P(CH2OH)4+, two resonance structures for the Wittig reagent Ph3PCH2, and pentaphenylphosphorane, a rare pentaorganophophorus compound.

The parent phosphorane (σ5λ5) is PH5, which is unknown.[citation needed] Related compounds containing both halide and organic substituents on phosphorus are fairly common. Those with five organic substituents are rare, although P(C6H5)5 is known, being derived from P(C6H5)4+ by reaction with phenyllithium.[citation needed]
Phosphorus ylides are unsaturated phosphoranes, known as Wittig reagents, e.g. CH2P(C6H5)3. These compounds feature tetrahedral phosphorus(V) and are considered relatives of phosphine oxides. They also are derived from phosphonium salts, but by deprotonation not alkylation.[citation needed]
Organophosphorus(III) compounds, main categories
[edit]Phosphites, phosphonites, and phosphinites
[edit]Phosphites, sometimes called phosphite esters, have the general structure P(OR)3 with oxidation state +3. Such species arise from the alcoholysis of phosphorus trichloride:
- PCl3 + 3 ROH → P(OR)3 + 3 HCl
The reaction is general, thus a vast number of such species are known. Phosphites are employed in the Perkow reaction and the Michaelis–Arbuzov reaction. They also serve as ligands in organometallic chemistry.
Intermediate between phosphites and phosphines are phosphonites (P(OR)2R') and phosphinite (P(OR)R'2). Such species arise via alcoholysis reactions of the corresponding phosphonous and phosphinous chlorides ((PCl2R') and (PClR'2) , respectively). The latter are produced by reaction of a phosphorus trichloride with a poor metal-alkyl complex, e.g. organomercury, organolead, or a mixed lithium-organoaluminum compound.[8]
Phosphines
[edit]The parent compound of the phosphines is PH3, called phosphine in the US and British Commonwealth, but phosphane elsewhere.[9] Replacement of one or more hydrogen centers by an organic substituents (alkyl, aryl), gives PH3−xRx, an organophosphine, generally referred to as phosphines.[citation needed]
From the commercial perspective, the most important phosphine is triphenylphosphine, several million kilograms being produced annually. It is prepared from the reaction of chlorobenzene, PCl3, and sodium.[6] Phosphines of a more specialized nature are usually prepared by other routes.[10] Phosphorus halides undergo nucleophilic displacement by organometallic reagents such as Grignard reagents. Organophosphines are nucleophiles and ligands. Two major applications are as reagents in the Wittig reaction and as supporting phosphine ligands in homogeneous catalysis.[citation needed]
Their nucleophilicity is evidenced by their reactions with alkyl halides to give phosphonium salts. Phosphines are nucleophilic catalysts in organic synthesis, e.g. the Rauhut–Currier reaction and Baylis-Hillman reaction. Phosphines are reducing agents, as illustrated in the Staudinger reduction for the conversion of organic azides to amines and in the Mitsunobu reaction for converting alcohols into esters. In these processes, the phosphine is oxidized to phosphorus(V). Phosphines have also been found to reduce activated carbonyl groups, for instance the reduction of an α-keto ester to an α-hydroxy ester.[11]
A few halophosphines are known, although phosphorus' strong nucleophilicity predisposes them to decomposition, and dimethylphosphinyl fluoride spontaneously disproportionates to dimethylphosphine trifluoride and tetramethylbiphosphine.[12] One common synthesis adds halogens to tetramethylbiphosphine disulfide.[13] Alternatively alkylation of phosphorus trichloride gives a halophosphonium cation, which metals reduce to halophosphines.
Phosphaalkenes and phosphaalkynes
[edit]Compounds with carbon phosphorus(III) multiple bonds are called phosphaalkenes (R2C=PR) and phosphaalkynes (RC≡P). They are similar in structure, but not in reactivity, to imines (R2C=NR) and nitriles (RC≡N), respectively. In the compound phosphorine, one carbon atom in benzene is replaced by phosphorus. Species of this type are relatively rare but for that reason are of interest to researchers. A general method for the synthesis of phosphaalkenes is by 1,2-elimination of suitable precursors, initiated thermally or by base such as DBU, DABCO, or triethylamine:

Thermolysis of Me2PH generates CH2=PMe, an unstable species in the condensed phase.
Organophosphorus(0), (I), and (II) compounds
[edit]Compounds where phosphorus exists in a formal oxidation state of less than III are uncommon, but examples are known for each class. Organophosphorus(0) species are debatably illustrated by the carbene adducts, [P(NHC)]2, where NHC is an N-heterocyclic carbene.[14] With the formulae (RP)n and (R2P)2, respectively, compounds of phosphorus(I) and (II) are generated by reduction of the related organophosphorus(III) chlorides:[citation needed]
Diphosphenes, with the formula R2P2, formally contain phosphorus-phosphorus double bonds. These phosphorus(I) species are rare but are stable provided that the organic substituents are large enough to prevent catenation. Bulky substituents also stabilize phosphorus radicals.
Many mixed-valence compounds are known, e.g. the cage P7(CH3)3.
See also
[edit]- Activity-based proteomics—A branch of biochemistry that often relies on organophosphorus probes to interrogate enzyme activities
- Bihar school meal poisoning incident
- Organophosphates
- Organophosphites
- Organothiophosphates
References
[edit]- ^ Merriam-Webster, Merriam-Webster's Unabridged Dictionary, Merriam-Webster, archived from the original on 2020-05-25, retrieved 2015-12-17.
- ^ Lewis, Robert Alan (1998). Lewisʼ Dictionary of Toxicology. CRC Lewis. p. 763. ISBN 978-1-56670-223-2. Retrieved 18 July 2013.
- ^ Dillon, K. B.; Mathey, F.; Nixon, J. F. (1997) Phosphorus. The Carbon Copy; John Wiley & Sons, ISBN 0-471-97360-2
- ^ Quin, L. D. (2000) A Guide to Organophosphorus Chemistry; John Wiley & Sons, ISBN 0-471-31824-8
- ^ Racke, K.D. (1992). "Degradation of organophosphorus insecticides in environmental matrices", pp. 47–73 in: Chambers, J.E., Levi, P.E. (eds.), Organophosphates: Chemistry, Fate, and Effects. Academic Press, San Diego, ISBN 0121673456.
- ^ a b c Svara, Jürgen; Weferling, Norbert & Hofmann, Thomas (2006). "Phosphorus Compounds, Organic". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a19_545.pub2. ISBN 978-3527306732.
- ^ a b c Almasi, Lucreţia (1971). "The Sulfur–Phosphorus Bond". In Senning, Alexander (ed.). Sulfur in Organic and Inorganic Chemistry. Vol. 1. New York: Marcel Dekker. pp. 39–106. ISBN 0-8247-1615-9. LCCN 70-154612.
- ^ Engel, Robert; Cohen, JaimeLee Iolani (2004). Synthesis of Carbon-Phosphorus Bonds (2 ed.). Boca Raton: CRC Press. §4.3.2.1–7. ISBN 0-8493-1617-0. LCCN 2003060796.
- ^ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "phosphanes". doi:10.1351/goldbook.P04548
- ^ Downing, J.H.; Smith, M.B. (2003). "Phosphorus Ligands". Comprehensive Coordination Chemistry II. 2003: 253–296. doi:10.1016/B0-08-043748-6/01049-5. ISBN 9780080437484.
- ^ Zhang, W.; Shi, M. (2006). "Reduction of activated carbonyl groups by alkyl phosphines: formation of α-hydroxy esters and ketones". Chem. Commun. 2006 (11): 1218–1220. doi:10.1039/b516467b. PMID 16518496.
- ^ Seel, F.; Rudolph, K. (1968) [12 March 1968]. "Über Dimethylfluorphosphin und Dimethyldifluorphosphoran". Z. anorg. allg. Chem. (in German). 359 (333): 233–244. doi:10.1002/zaac.19683630503.
- ^ Durig, J. R.; Saunders, J. E. (1975) [25 Oct 1974]. "Spectra and structure of organophosphorus compounds XI: Infrared and Raman spectra of (CH3)2PCl". Journal of Molecular Structure. 27. Elsevier: 403–416. doi:10.1016/0022-2860(75)87051-7. Adapted from Saunders, J. E. (August 1972). (PhD). University of South Carolina.
{{cite thesis}}: Missing or empty|title=(help) - ^ Wang, Yuzhong; Xie, Yaoming; Wei, Pingrong; King, R. Bruce; Schaefer, Iii; Schleyer, Paul v. R.; Robinson, Gregory H. (2008). "Carbene-Stabilized Diphosphorus". Journal of the American Chemical Society. 130 (45): 14970–1. doi:10.1021/ja807828t. PMID 18937460.
External links
[edit]- Organophosphorus chemistry at users.ox.ac.uk
- Organophosphorus chemistry at www.chem.wisc.edu
- NMR predictor for organophosphorus compound chemical shifts from Alan Brisdon's Research Group
Compounds of carbon with other elements in the periodic table | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Legend |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||